
Boală renală polichistică autosomală dominantă - Autosomal dominant polycystic kidney disease
| Boală renală polichistică autosomală dominantă | |
|---|---|
| Alte nume | PKD autosomal dominant, PKD cu debut la adulți |
 | |
| Rinichii polichistici | |
| Specialitate |
Genetica medicală |
Boala renală polichistică autosomală dominantă ( ADPKD ) este cea mai răspândită, potențial letală, tulburare umană monogenă . Este asociat cu o mare variabilitate interfamilială și intrafamilială, care poate fi explicată în mare măsură prin eterogenitatea sa genetică și genele modificatoare . Este, de asemenea, cea mai frecventă dintre afecțiunile renale chistice moștenite - un grup de tulburări cu patogeneză conexă, dar distinctă, caracterizată prin dezvoltarea chisturilor renale și a diverselor manifestări extrarenale, care în cazul ADPKD includ chisturi în alte organe, cum ar fi ficatul , vezicule seminale , pancreas și membrană arahnoidă , precum și alte anomalii, cum ar fi anevrisme intracraniene și dolicoectazii , dilatarea și anevrismele rădăcinii aortice , prolapsul valvei mitrale și herniile peretelui abdominal . Peste 50% dintre pacienții cu ADPKD dezvoltă în cele din urmă boli de rinichi în stadiul final și necesită dializă sau transplant de rinichi . Se estimează că ADPKD afectează cel puțin unul din 1000 de indivizi din întreaga lume, ceea ce face ca această boală să fie cea mai frecventă afecțiune renală moștenită cu o prevalență diagnosticată de 1: 2000 și o incidență de 1: 3000-1: 8000 la scară globală.
semne si simptome
Printre prezentările clinice se numără:
- Dureri acute de lomb
- Sânge în urină
- Rinichi votabili
- Hemoragie subarahnoidiană (anevrism de boabe)
- Hipertensiune
- Chisturi hepatice asociate
- Uremie datorată insuficienței renale
- Anemie datorată bolilor renale cronice
- Creșteți secreția eritrocitelor sau eritropoietinei
Genetica
ADPKD este eterogen genetic, cu două gene identificate: PKD1 (regiunea cromozomială 16p13.3; aproximativ 85% cazuri) și PKD2 (4q21; aproximativ 15% cazuri). Mai multe mecanisme genetice contribuie probabil la exprimarea fenotipică a bolii. Deși există dovezi pentru un mecanism cu două lovituri (linia germinativă și inactivarea somatică a două alele PKD) care explică dezvoltarea focală a chisturilor renale și hepatice, haploinsuficiența este mai probabil să explice manifestările vasculare ale bolii. În plus, noile modele de șoareci homozigote pentru alelele hipomorfe PKD1 22 și 23 și demonstrarea proliferării crescute a celulelor epiteliale renale la șoareci PKD2 +/− sugerează că alte mecanisme decât ipoteza cu două lovituri contribuie, de asemenea, la fenotipul chistic.
În ADPKD apare o mare variabilitate interfamilială și intrafamilială. Majoritatea indivizilor cu mutații PKD1 au insuficiență renală până la vârsta de 70 de ani, în timp ce mai mult de 50% dintre indivizii cu mutații PKD2 au funcție renală adecvată la acea vârstă (vârsta medie de debut a bolii renale în stadiul final: 54,3 ani cu PKD1 ; 74 · 0 ani cu PKD2 ).
Variabilitatea intrafamilială semnificativă observată în severitatea manifestărilor renale și extrarenale indică factori de modificare genetică și de mediu care pot influența rezultatul ADPKD, iar rezultatele unei analize a variabilității funcției renale între gemenii monozigoți și frați susțin rolul modificatorilor genetici. în această boală. Se estimează că 43-78% din varianța vârstei față de ESRD s-ar putea datora factorilor modificatori ereditari, părinții la fel de probabil ca și copiii să prezinte o boală mai severă în studiile de perechi părinte-copil.
Fiziopatologie
La mulți pacienți cu ADPKD, disfuncția renală nu este evidentă clinic până la 40 sau 50 de ani de viață. Cu toate acestea, un număr tot mai mare de dovezi sugerează că formarea chisturilor renale începe în uter . Chisturile se formează inițial ca mici dilatații în tubulii renali, care apoi se extind formând cavități umplute cu lichide de diferite dimensiuni. Factorii sugerați să conducă la cistogeneza includ o mutație a liniei germinale într-una dintre alelele genei policistinei, un al doilea impact somatic care duce la pierderea alelei normale și un al treilea impact, care poate fi o insultă renală care declanșează proliferarea celulară și o răspuns la vătămare. . Datorită numeroaselor similitudini între fiziopatologia ADPKD și fiziopatologia răspunsului renal la leziuni, ADPKD a fost descrisă ca o stare de activare aberantă și persistentă a căilor de răspuns la leziuni renale. În progresia bolii, dilatarea continuă a tubulilor prin proliferarea crescută a celulelor, secreția de lichide și separarea de tubul parental duc la formarea chisturilor.
ADPKD, împreună cu multe alte boli care prezintă chisturi renale, pot fi clasificate într-o familie de boli cunoscute sub numele de ciliopatii . Celulele epiteliale ale tubulilor renali, inclusiv toate segmentele nefronului și ale canalelor colectoare (cu excepția celulelor intercalate) prezintă prezența unui singur cilium apical primar. Polichistina-1 , proteina codificată de gena PKD1 , este prezentă pe acești cili și se crede că simte fluxul cu domeniile sale extracelulare mari, activând canalele de calciu asociate cu polichistina-2 , produsul genei PKD2 , ca urmare a setarea genetică a ADPKD așa cum este explicată în subsecțiunea genetică de mai sus.
Proliferarea celulelor epiteliale și secreția de lichide care duc la cistogeneza sunt două caracteristici distinctive în ADPKD. În primele etape ale cistogenezei, chisturile sunt atașate de tubulii renali parentali și un derivat al filtratului glomerular pătrunde în chisturi. Odată ce aceste chisturi se extind la aproximativ 2 mm în diametru, chistul se închide din tubul său parental și, după aceea, lichidul poate pătrunde în chisturi numai prin secreția transepitelială, care la rândul său este sugerată să crească datorită efectelor secundare de la o concentrație intracelulară crescută a ciclului AMP (AMPc).
Din punct de vedere clinic, creșterea insidioasă a numărului și dimensiunii chisturilor renale se traduce ca o creștere progresivă a volumului renal. Studiile conduse de profesioniștii Clinicii Mayo au stabilit că volumul total al rinichilor (TKV) într-o cohortă mare de pacienți cu ADPKD a fost de 1060 ± 642 ml, cu o creștere medie de 204 ml pe parcursul a trei ani, sau 5,27% pe an în cursul natural al bolii, printre alte descoperiri importante, noi, care au fost studiate pe larg pentru prima dată.

Diagnostic
De obicei, diagnosticul ADPKD se realizează inițial prin imagistica renală utilizând ultrasunete , tomografie sau RMN . Cu toate acestea, diagnosticarea moleculară poate fi necesară în următoarele situații: 1- când este necesar un diagnostic clar la indivizii tineri, cum ar fi un potențial donator legat de viață într-o familie afectată cu date echivoce de imagistică; 2- la pacienții cu antecedente familiale negative de ADPKD, din cauza potențialei suprapuneri fenotipice cu alte câteva boli chistice renale; 3- la familiile afectate de boală renală polichistică cu debut precoce, deoarece în acest caz pot fi implicate alele hipomorfe și / sau moștenirea oligogenă ; și 4- la pacienții care solicită consiliere genetică , în special la cuplurile care doresc un diagnostic genetic pre-implantare .
Constatările rinichilor ecogeni mari fără chisturi macroscopice distincte la un sugar / copil cu un risc de 50% pentru ADPKD sunt diagnostice. În absența unui istoric familial de ADPKD, prezența măririi și chisturilor renale bilaterale, cu sau fără prezența chisturilor hepatice și absența altor manifestări sugestive ale unei boli chistice renale diferite oferă dovezi prezumtive, dar nu definite, pentru diagnosticul. În unele cazuri, anevrismele intracraniene pot fi un semn asociat al ADPKD, iar screening-ul poate fi recomandat pacienților cu antecedente familiale de anevrism intracranian.
Testarea genetică moleculară prin analiza legăturii sau screeningul mutației directe este disponibil clinic; cu toate acestea, eterogenitatea genetică este o complicație semnificativă la testarea genetică moleculară . Uneori, un număr relativ mare de membri ai familiei afectați trebuie testați pentru a stabili care dintre cele două gene posibile este responsabilă în cadrul fiecărei familii. Dimensiunea mare și complexitatea genelor PKD1 și PKD2 , precum și heterogenitatea alelică marcată , prezintă obstacole în calea testării moleculare prin analiza directă a ADN-ului . Sensibilitatea testării este de aproape 100% pentru toți pacienții cu ADPKD care au vârsta de 30 de ani sau mai mult și pentru pacienții mai tineri cu mutații PKD1 ; aceste criterii sunt sensibile doar la 67% pentru pacienții cu mutații PKD2 ]] care sunt mai mici de 30 de ani.

Rinichi polichistic adult

Diagrama bolii polichistice autosomale dominante cu insetare renală normală pentru comparație
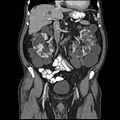
Scanare computerizată abdominală a unui adult cu boală renală polichistică autosomală dominantă: Formarea extinsă de chist se observă pe ambii rinichi, cu câteva chisturi și în ficat. ( Plan coronar )



Tratament
În prezent, singurul tratament farmacologic disponibil pentru ADPKD constă în reducerea vitezei de creștere a volumului renal total (TKV) cu antagoniști ai receptorului vasopresinei 2 (V2) (adică tolvaptan). Tratamentul cu tolvaptan nu oprește sau inversează progresia bolii și pacienții continuă să progreseze spre insuficiență renală. Modalitățile de tratament paliativ implică medicamente simptomatice (analgezice nonopioide și opioide) pentru durerea abdominală / retroperitoneală. Opțiunile pentru durerea rezistentă la analgezice includ proceduri chirurgicale simple sau complexe (de exemplu, aspirația chistului renal, decorticarea chistului, denervarea renală și nefrectomia), care pot duce la complicații inerente intervenției chirurgicale. Cercetări recente sugerează că intervențiile dietetice ketogene afectează în mod benefic progresia și simptomele la persoanele cu ADPKD. Pierderea ușoară în greutate afectează favorabil durerea, indicând beneficiile modificărilor dietetice și ale stilului de viață.
Medicație acvaretică
În 2014, Japonia a fost prima țară din lume care a aprobat un tratament farmacologic pentru ADPKD urmat de Canada și Europa, care a aprobat medicamentul tolvaptan pentru pacienții cu ADPKD la începutul anului 2015. FDA SUA a aprobat utilizarea tolvaptanului în tratamentul ADPKD în 2018. Tolvaptanul, un aquaretic medicament, este un receptor de vasopresină 2 (V2) antagonist . Studiile preclinice au sugerat că molecula AMPc ar putea fi implicată în extinderea chisturilor ADPKD, iar studiile pe rozătoare au confirmat rolul vasopresinei în creșterea nivelului AMPc la rinichi, care a pus bazele conducerii studiilor clinice. Deoarece datele din Consortium for Radiologic Imaging Studies of Polycystic Kidney Disease (CRISP) conduse de Mayo Clinic au arătat că volumul total de rinichi (TKV) a prezis riscul apariției bolii renale cronice la pacienții cu ADPKD, studiul TEMPO 3: 4, care a înscris pacienții din 129 de site-uri din întreaga lume, din 2007 până în 2009, au evaluat TKV ca principal obiectiv final pentru a testa eficacitatea tolvaptanului la pacienții cu ADPKD. Studiul respectiv a arătat o scădere semnificativă a raportului dintre creșterea TKV și descurajarea scăderii funcției renale la pacienții cu ADPKD după tratamentul cu tolvaptan; cu toate acestea, deoarece rezultatele testelor de laborator privind funcția hepatică au apărut crescute la un procent de pacienți înscriși în acel studiu, aprobarea medicamentului a fost fie întârziată de agențiile de reglementare, fie, ca în cazul SUA, a fost total negată.
Intervenții dietetice și de viață
Cercetările folosind modele ADPKD de șoarece au arătat că restricția ușoară a alimentelor a îmbunătățit puternic progresia bolii. S-a demonstrat că mecanismul implică starea metabolică a cetozei, iar efectele benefice ar putea fi produse prin hrănirea cu timp limitat, postul acut, o dietă ketogenică sau prin suplimentarea cu cetonă beta-hidroxibutirat la modelele ADPKD de șoarece, șobolan și pisică. Un regim de dietă ketogenică nu numai că a oprit progresia ulterioară a bolii, dar a condus la inversarea parțială a bolii chistice renale la un model de șobolan. Starea metabolică a cetozei poate fi benefică în ADPKD, deoarece celulele chistului renal din ADPKD prezintă un defect metabolic similar cu efectul Warburg în cancerul care le face foarte dependente de glucoză și incapabile să metabolizeze acizii grași și cetonele. În concordanță cu aceasta, nivelurile serice de glucoză se corelează pozitiv cu progresia mai rapidă a bolii la pacienții cu ADPKD. De asemenea, indivizii cu ADPKD și diabetul de tip 2 au un volum renal total (TKV) semnificativ mai mare decât cei cu ADPKD singură, iar supraponderalitatea sau obezitatea se asociază cu o progresie mai rapidă în ADPKD în stadiu incipient. Un studiu retrospectiv din seria de cazuri a arătat că simptomele bolii ADPKD - inclusiv durerea, hipertensiunea și funcția renală - s-au îmbunătățit la 131 de pacienți care au implementat diete ketogenice pe o durată medie de 6 luni.
Aportul alimentar de sodiu este asociat cu o scădere mai gravă a funcției renale în ADPKD, iar limitarea aportului de sodiu este recomandată în general pacienților. Nu s-a constatat că aportul de proteine din dietă se corelează cu progresia ADPKD.
Creșterea aportului de apă este considerată benefică în ADPKD și este recomandată în general. Mecanismul benefic care stă la baza creșterii aportului de apă poate fi legat de efectele asupra receptorului vasopresinei V2 sau se poate datora suprimării formării dăunătoare de microcristale din tubulii renali prin diluarea substanțelor dizolvate precum oxalat de calciu, fosfat de calciu și acid uric.
Medicație analgezică
Durerea cronică la pacienții cu ADPKD este adesea refractară la tratamentele conservatoare, neinvazive, dar analgezicele nonopioide și intervențiile conservatoare pot fi utilizate mai întâi înainte de a lua în considerare analgezicele opioide ; dacă durerea continuă, atunci intervențiile chirurgicale pot viza chisturile renale sau hepatice pentru a aborda direct cauza durerii, cu opțiuni chirurgicale incluzând decorticarea chistului renal , denervarea renală și nefrectomia .
Aspirația chistului renal
Aspirarea cu scleroterapie cu etanol poate fi efectuată pentru tratamentul chisturilor renale simple simptomatice, dar poate fi impracticabilă la pacienții avansați cu chisturi multiple. Procedura în sine constă în introducerea percutană a unui ac în chistul identificat, sub îndrumare cu ultrasunete , cu scurgerea ulterioară a lichidului conținut; scleroterapia este utilizată pentru a evita reacumularea lichidă care poate apărea în chist, ceea ce poate duce la reapariția simptomelor.
Decorticarea laparoscopică a chistului
Decorticarea laparoscopică a chistului (denumită și marsupializare) constă în îndepărtarea unuia sau mai multor chisturi renale prin intervenții chirurgicale laparoscopice , în timpul cărora chisturile sunt perforate, iar peretele exterior al chisturilor mai mari este excizat cu grijă pentru a nu inciza parenchimul renal. Această procedură poate fi utilă pentru ameliorarea durerii la pacienții cu ADPKD și este de obicei indicată după ce aspirația anterioară a chistului a confirmat că chistul care trebuie decorticat este responsabil pentru durere. Studiile controlate non-randomizate efectuate în anii '90 au arătat că pacienții cu chisturi renale simple simptomatice care au recidivat simptomele după răspunsul inițial la aspirație simplă pot fi supuși în siguranță la decorticarea chistului, cu o durată medie de viață fără durere între 17 și 24 de luni după operație. Decorticarea laparoscopică prezintă o rată de recurență de 5% a chisturilor renale comparativ cu o rată de recurență de 82% obținută cu scleroterapia.
Neuroliză
Un tratament nou al durerii cronice suferite de mulți bolnavi de ADPKD este neuroliza plexului celiac . Aceasta implică ablația chimică a plexului celiac , pentru a provoca o degenerare temporară a fibrelor nervoase vizate. Atunci când fibrele nervoase degenerează, provoacă o întrerupere a transmiterii semnalelor nervoase. Acest tratament, când are succes, oferă o ameliorare semnificativă a durerii pentru o perioadă cuprinsă între câteva zile și peste un an. Procedura poate fi repetată atunci când nervii afectați s-au vindecat și durerea revine.
Nefrectomie
Mulți pacienți cu ADPKD suferă sechele simptomatice ca urmare a bolii, cum ar fi hemoragia chistului , durerea de flanc , infecțiile recurente , nefrolitiaza și simptomele efectului de masă (de exemplu, sațietate precoce , greață și vărsături și disconfort abdominal), din rinichii lor măriți. În astfel de cazuri, nefrectomia poate fi necesară datorită simptomelor intratabile sau când, în cursul pregătirii pentru transplantul de rinichi , rinichii nativi afectează adevăratul pelvis și exclud plasarea unei alogrefe donatoare . În plus, nefrectomia nativă poate fi efectuată în prezența suspiciunii de malignitate, deoarece carcinomul cu celule renale (RCC) este de două până la trei ori mai probabil în populația ADPKD din boala renală în stadiu final (ESKD) decât la pacienții cu ESKD fără ADPKD. Deși indicațiile pentru nefrectomie în ADPKD pot fi legate de mărimea rinichilor, decizia de a continua nefrectomia nativă este adesea luată individual, fără referințe specifice la măsurători ale dimensiunii rinichilor.
Dializă
Două modalități de dializă pot fi utilizate în tratamentul pacienților cu ADPKD: dializa peritoneală și hemodializa . Datele epidemiologice arată că ADPKD afectează 5-13,4% dintre pacienții supuși hemodializei în Europa și Statele Unite și aproximativ 3% în Japonia. Dializa peritoneală a fost de obicei contraindicată la pacienții cu ADPKD cu volum mare de rinichi și ficat, din cauza dificultăților fizice preconizate în procedură și a posibilelor complicații; cu toate acestea, nu se observă nicio diferență în morbiditatea pe termen lung între hemodializă și dializa peritoneală în ADPKD.
Transplant de rinichi
Transplantul de rinichi este acceptat ca tratament preferat pentru pacienții cu ADPKD cu ESRD. Dintre pacienții americani aflați pe lista de așteptare pentru transplant de rinichi (începând cu decembrie 2011), 7256 (8,4%) au fost enumerate din cauza bolii renale chistice și din cele 16 055 de transplanturi renale efectuate în 2011, 2057 (12,8%) au fost efectuate pentru pacienții cu chist boli de rinichi, cu 1.189 de la donatori decedați și 868 de la donatori vii.
Prognoză
La pacienții cu ADPKD, dezvoltarea și extinderea treptată a chistului duc la mărirea rinichilor și, în cursul bolii, rata filtrării glomerulare rămâne normală timp de decenii înainte ca funcția renală să înceapă să se deterioreze progresiv, ceea ce face dificilă predicția timpurie a rezultatului renal. Studiul CRISP, menționat în secțiunea de tratament de mai sus, a contribuit la construirea unei rațiuni solide care să susțină valoarea prognostică a volumului renal total (TKV) în ADPKD; TKV (evaluat prin RMN ) crește constant și o rată mai mare de mărire a rinichilor corelată cu scăderea accelerată a RFG, în timp ce TKV (HtTKV) ajustat pe înălțime al pacientului ≥600 ml / m prezice dezvoltarea bolii renale cronice în stadiul 3 în decurs de 8 ani.
Pe lângă TKV și HtTKV, rata estimată de filtrare glomerulară (eGFR) a fost, de asemenea, tentativ utilizată pentru a prezice progresia ADPKD. După analiza CT sau RMN a 590 de pacienți cu ADPKD tratați la Centrul de boli renale polichistice translaționale Mayo , Irazabal și colegii au dezvoltat un sistem de clasificare bazat pe imagistică pentru a prezice rata declinului eGFR la pacienții cu ADPKD. În această metodă de prognostic, pacienții sunt împărțiți în cinci subclase ale ratelor de creștere renală estimate în funcție de intervalele HtTKV specifice vârstei (1A, <1,5%; 1B, 1,5-3,0%; 1C, 3,0-4,5%; 1D, 4,5-6,0% și 1E,> 6,0%), așa cum este definit în studiul CRISP. Declinul eGFR în anii care urmează măsurării inițiale a TKV este semnificativ diferit între toate cele cinci subclase de pacienți, cei din subclasa 1E având cel mai rapid declin. Unele dintre cele mai frecvente cauze de deces la pacienții cu ADPKD sunt diverse infecții (25%), anevrismul de boabe rupt (15%) sau boala cardiacă coronariană / hipertensivă (40%).
Referințe
linkuri externe
- https://web.archive.org/web/20110608142128/http://kidney.niddk.nih.gov/kudiseases/pubs/polycystic/index.htm
- https://www.ncbi.nlm.nih.gov/disease/PKD.html
| Clasificare | |
|---|---|
| Resurse externe |